In 2023, Metascape introduced AI-generated summaries for gene functions and disease implications [see previous Blog]. Powered by ChatGPT-3 at the time, these summaries served as a highly beneficial complement to NCBI’s human annotations by filling critical gaps in disease context.
In our latest database update, we are thrilled to release a completely revamped annotation dataset generated using ChatGPT-5 via Copilot. The leap in quality, scientific precision, and clinical relevance is remarkable.
To validate this upgrade, we manually reviewed multiple gene entries across the spectrum — from heavily studied genes like TP53 to lesser-known targets like CCDC74A. We also employed Google Gemini as an independent AI reviewer to compare the generations in a blind test. The result? The new version wins 100% of the time.
Please enjoy this valuable contribution of AI development to our biomedical research community. Below are two examples highlighting the dramatic improvements you can expect to see.
Example 1: TP53 (A Well-Understood Gene)
Even for highly characterized genes with mature 2023 annotations, the new version delivers significant upgrades in nuance, structure, and clinical accuracy.
The 2023 Version (ChatGPT-3)
Function: The TP53 gene encodes a tumor suppressor protein that responds to various cellular stresses to regulate the expression of target genes, inducing cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. The protein contains transcriptional activation, DNA binding, and oligomerization domains. Mutations in this gene are associated with various human cancers, including hereditary cancers such as Li-Fraumeni syndrome. Alternative splicing and the use of alternate promoters result in multiple transcript variants and isoforms. Additional isoforms have also been shown to result from the use of alternate translation initiation codons from identical transcript variants.
Disease: The TP53 gene is one of the most frequently mutated genes in human cancers, with mutations occurring in over 50% of all tumors. These mutations can lead to loss of function of the TP53 protein, resulting in uncontrolled cell growth and tumor formation. As a result, there has been significant interest in developing drugs that can restore TP53 function or target tumors with TP53 mutations. One example of a successful drug targeting TP53 mutations is the small molecule PRIMA-1, which has been shown to restore TP53 function in vitro and in vivo. Another promising approach is the use of gene therapy to introduce functional TP53 into tumor cells. Several clinical trials are currently underway to evaluate the safety and efficacy of these approaches in cancer patients.
The New Version (ChatGPT-5)
Function: TP53 encodes the tumor suppressor p53, a transcription factor that safeguards genomic integrity. In response to DNA damage, oncogene activation, hypoxia, or other stresses, p53 activates or represses target genes controlling cell cycle arrest, apoptosis, senescence, DNA repair, and metabolism. It functions as a sequence-specific DNA-binding tetramer with transactivation and oligomerization domains, integrating diverse stress signals into coordinated cellular outcomes. Alternative isoforms and post-translational modifications fine-tune p53 activity across tissues and contexts. Because of its central role in preventing malignant transformation, TP53 is often called the “guardian of the genome.”
Disease: TP53 is the most frequently mutated gene in human cancer, with loss-of-function or dominant-negative mutations driving genomic instability, therapy resistance, and aggressive tumor behavior. Germline TP53 mutations cause Li-Fraumeni syndrome, predisposing carriers to early-onset sarcomas, breast cancer, brain tumors, and leukemias. Drug discovery has focused on restoring or exploiting p53 pathways, including MDM2 inhibitors (e.g., idasanutlin in trials) that stabilize wild-type p53, and small molecules like eprenetapopt (APR-246) designed to refold certain mutant p53 proteins. While no universal p53-restoring drug is approved, many standard chemotherapies and radiotherapy rely on p53-mediated cell death, and p53 pathway modulators remain a major oncology research frontier.
Google Gemini’s Review:
Verdict: The New Version (was provided as Version B) is clearly superior.
Version B offers a more precise, scientifically rigorous narrative, rightly highlighting p53 as the “guardian of the genome” and explaining its structural function as a tetramer. In the disease section, version B provides critical clinical specifics, explicitly detailing the cancers associated with Li-Fraumeni syndrome. Furthermore, its therapeutic landscape is much more up-to-date and nuanced. Instead of broadly referencing older compounds like PRIMA-1 or generic gene therapy, it accurately cites modern clinical targets like MDM2 inhibitors (idasanutlin) and advanced refolding molecules (eprenetapopt).
Example 2: CCDC74A (A Lesser-Understood Gene)
For newly discovered or less-characterized genes, the older model often struggled with organization and relied on vague generalizations. The new model connects structural motifs directly to functional biology.
The 2023 Version (ChatGPT-3)
There is limited information available on targeted drug discovery efforts for CCDC74A, as it is a relatively newly discovered gene. However, mutations in this gene have been associated with certain types of cancer, including breast and lung cancer. This suggests that targeting CCDC74A may be a potential strategy for cancer treatment. Currently, there are no drugs on the market that specifically target CCDC74A. However, there are several drugs that target other proteins involved in cell division, such as mitotic inhibitors and cyclin-dependent kinase inhibitors, which may indirectly affect CCDC74A function. Further research is needed to fully understand the role of CCDC74A in cancer and to develop targeted therapies for this gene.
CCDC74A is a human gene that encodes for a protein called coiled-coil domain containing 74A. This protein is involved in the regulation of cell division and is expressed in various tissues throughout the body. Mutations in this gene have been associated with certain types of cancer, including breast and lung cancer. Additionally, CCDC74A has been shown to interact with other proteins involved in cell division, suggesting that it may play a role in the development and progression of cancer. Further research is needed to fully understand the function of CCDC74A and its potential as a therapeutic target for cancer treatment.
The New Version (ChatGPT-5)
CCDC74A encodes a coiled-coil domain-containing protein, a structural motif commonly involved in oligomerization and assembly of cytoskeletal or scaffolding complexes. Although its precise molecular partners are not fully defined, CCDC74A localizes to centrosomal or spindle-associated regions in some studies, suggesting a role in mitotic spindle organization, chromosome segregation, or microtubule dynamics. By contributing to the architecture of protein complexes that coordinate cell division, CCDC74A may influence proliferation, genome stability, and cell fate decisions in dividing cells.
The disease implications of CCDC74A are still being uncovered, but altered expression has been reported in certain cancers, where it may support uncontrolled proliferation or chromosomal instability. Disruption of spindle- or centrosome-associated proteins often leads to aneuploidy and tumor progression, making CCDC74A a potential contributor to oncogenic cell cycle phenotypes. No approved drugs directly target CCDC74A, yet tumors that depend on its function may be particularly sensitive to microtubule-targeting agents or mitotic checkpoint inhibitors already in clinical use. As functional genomics clarifies its role, CCDC74A could emerge as a biomarker or vulnerability in specific cancer subtypes.
Google Gemini’s Review:
Verdict: The New Version (was provided as version A) is far superior.
It provides a well-structured, scientifically precise explanation, offering specific mechanistic insights into CCDC74A’s role in centrosomal localization, spindle organization, and microtubule dynamics. Instead of relying on vague statements about “cell division,” version A uses accurate terminology to connect the protein’s coiled-coil structural motif to its functional biology. Furthermore, verison B is poorly organized—placing drug discovery before basic function and repeating the exact same point about breast and lung cancer in both paragraphs. Version A flows logically from molecular function to disease implications, correctly identifying how chromosomal instability relates to existing microtubule-targeting therapies.
A Decade of Impact, Innovation, and Community Growth in Functional Genomics
We are delighted to announce that Metascape, published in Nature Communications, has achieved a significant milestone — surpassing 10,000 citations. This remarkable accomplishment reflects Metascape’s lasting scientific impact and the ongoing support from the global bioinformatics community over the past decade.
From a Simple Idea to a Widely Used Platform
Metascape was launched in October 2015 with a clear mission: to give biologists an intuitive, accessible tool for functional enrichment analysis of gene lists. At the time, many enrichment tools required substantial computational expertise, creating barriers for experimental scientists. Metascape’s design lowered those barriers, making biological interpretation faster and more approachable for users at all levels.
As the community’s needs evolved, Metascape did too. Support for multiple gene lists enabled comparative analyses, while protein-protein interaction network analysis connected gene-level findings to broader biological systems. Over time, organism coverage expanded, backend databases were refined, and annotation quality improved, steadily transforming Metascape into a comprehensive platform for functional genomics.
The publication of Metascape in Nature Communications in April 2019 marked a major turning point, significantly increasing its visibility and adoption worldwide. Notably, dissemination through Chinese social media platforms such as WeChat played a crucial role in introducing Metascape to a broad community of researchers across China, accelerating its uptake and impact.
Measuring Impact: Citations and Community Reach
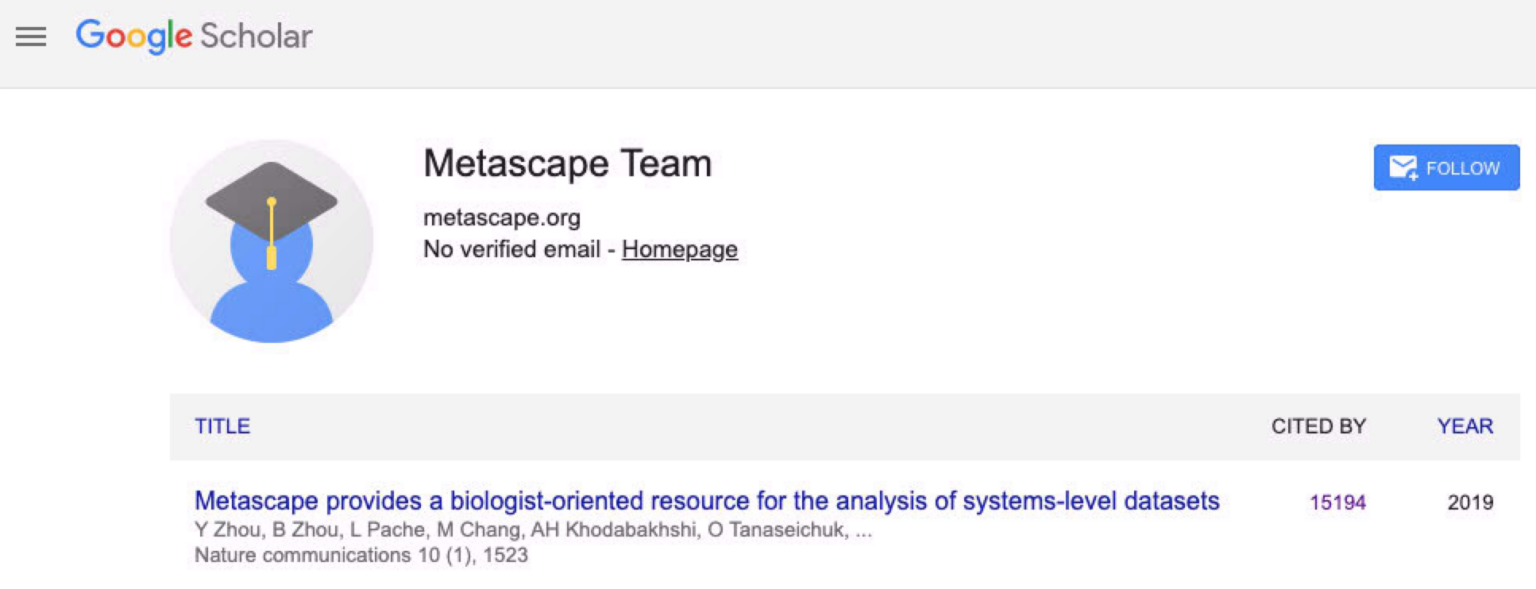
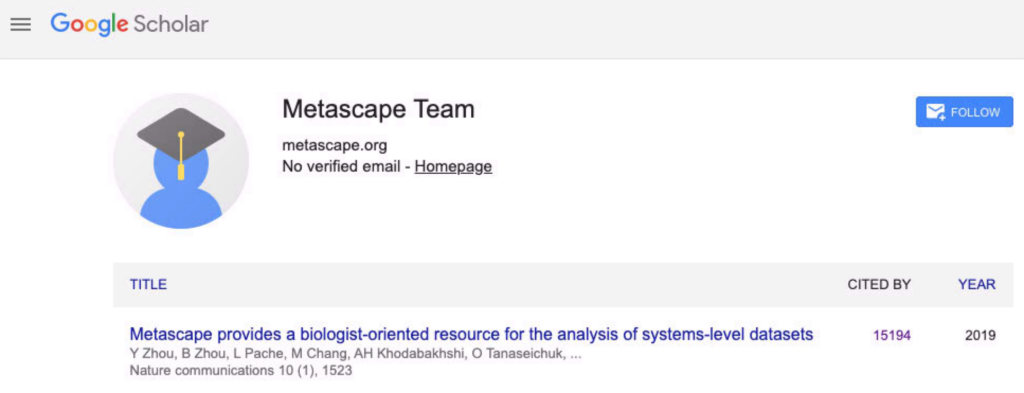
Today, Google Scholar reports over 15,000 citations for the Metascape paper.
A screenshot taken from Google Scholar on Mar 29, 2026.
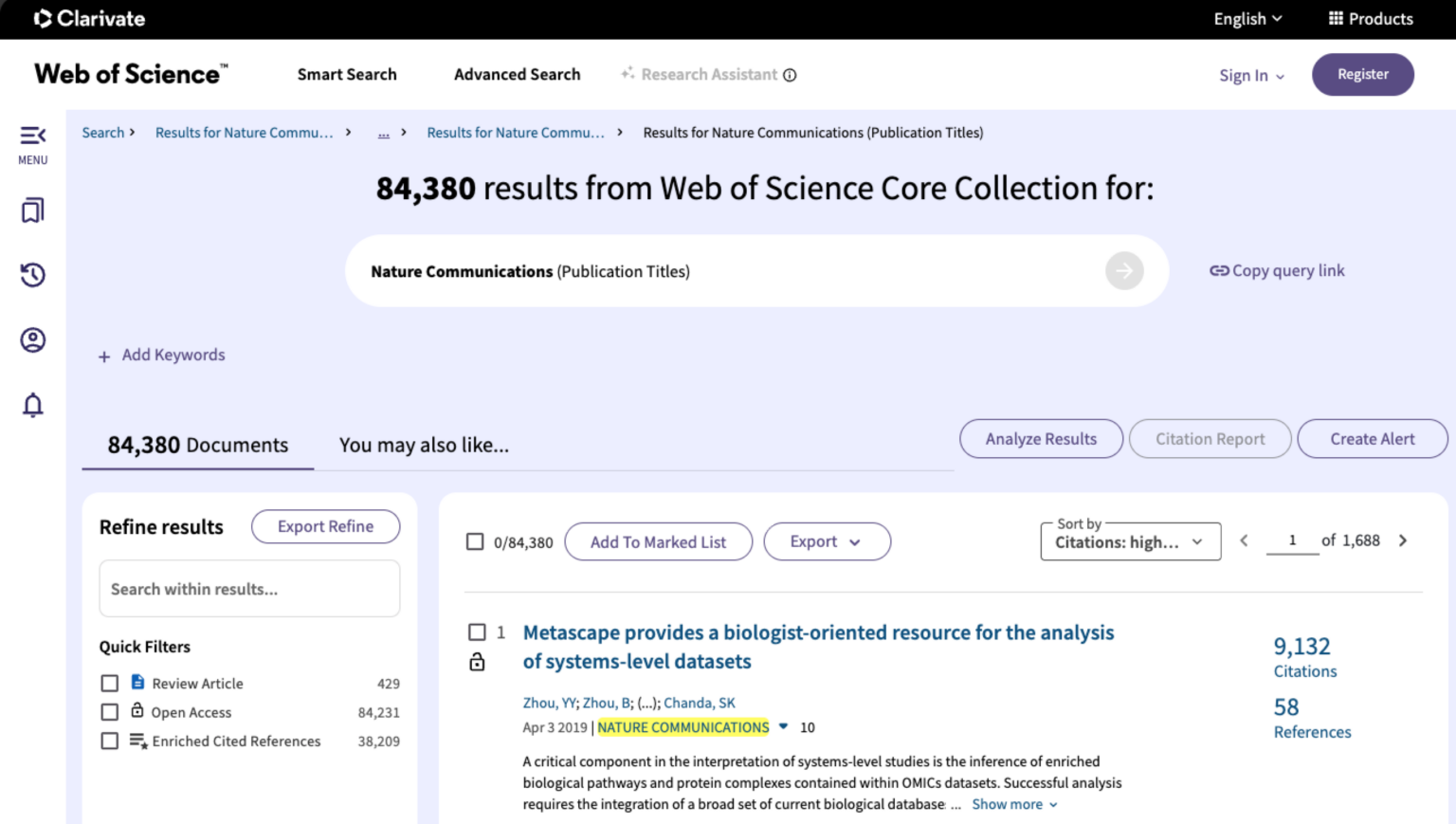
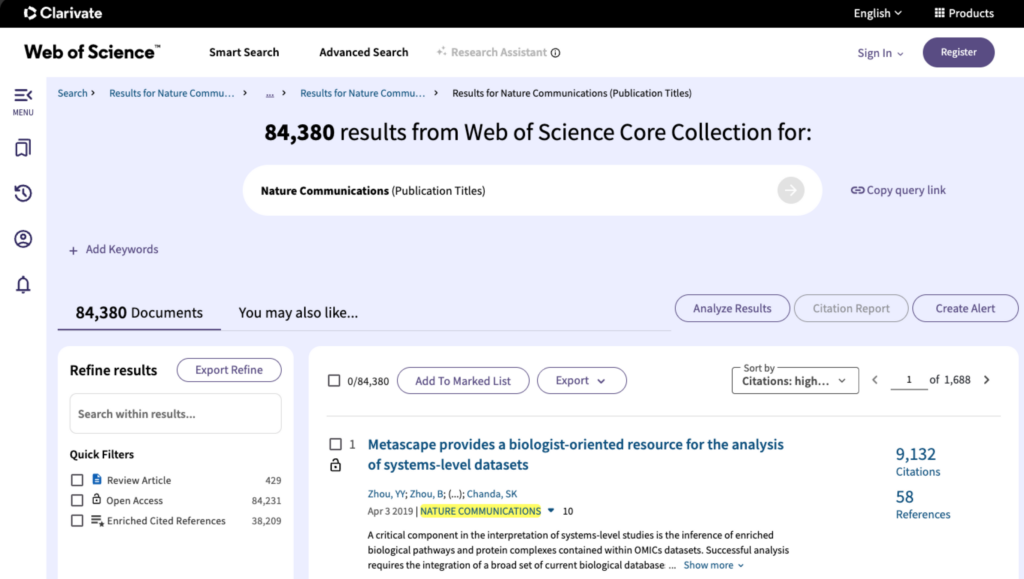
According to Web of Science, it is now the most cited article published in Nature Communications — a testament to its scientific relevance and practical utility.
A screenshot taken from https://www.webofscience.com on Mar 29, 2026.
We owe much of Metascape’s success to influential WeChat channels and bioinformatics content creators who have introduced Metascape to researchers throughout China. Their efforts have fostered widespread awareness, adoption, and community-driven learning, helping Metascape become a global standard in functional genomics analysis.
Continuous Innovation Behind a Stable Interface
Metascape’s enduring success is rooted in its philosophy of continual development paired with a stable, user-friendly interface. While the main interface has remained familiar and reliable, significant advancements have occurred behind the scenes. Notable milestones include MSBio (2021), which expanded multi-omics and biological interpretation capabilities, and the integration of ChatGPT-based human gene annotations (2023), offering AI-assisted, natural-language summaries to make gene-level knowledge more accessible and interpretable.
Volunteer-driven engineering efforts have prioritized infrastructure and reliability. Important improvements such as migration to AWS Cloud, robust server scaling, and ongoing performance optimizations ensure Metascape remains responsive and dependable as its user base grows. A dedicated team of volunteers consistently maintains and enhances the service, with around 50 Git commits per year underscoring steady backend evolution and long-term commitment. As demand increases, the team’s diligence ensures stability, addresses technical challenges, and responds to user feedback.
Looking Ahead
Metascape thrives thanks to its dedicated users, whose trust and feedback continually drive our progress. As we celebrate this milestone, we remain committed to providing a trusted, high-quality tool for functional enrichment and discovery. Thank you to all who have contributed — here’s to the next chapter!
Posted inComment, News|TaggedNews|Comments Off on Celebrating a Milestone: Metascape Surpasses 10,000 Citations
In the early days of natural language processing (NLP) research, we quantified the similarity between documents by counting shared words. However, this simplistic approach ignored synonyms, treating “cat” and “kitty” as distinct entities, much like “cat” and “rock.” (Figure 1. Left) The resulting identity-based word representation, known as one-hot encoding, fell short when applied to clustering documents. Fortunately, the NLP landscape has evolved, thanks to deep learning techniques like Word2Vec [1]. “cat” and “kitty” — once distant, now recognized as near identical due to Word2Vec (Figure 1. Right).
Figure 1. Left, in one-hot encoding, “cat” and “kitty” is as distant as “cat” and “rock”. The representation is unaware that “cat” and “kitty” are synonyms. Right, in Word2Vec representation, “cat” and “kitty” are very close and both are distant from “rock”, which correctly depicts the meanings of these words.
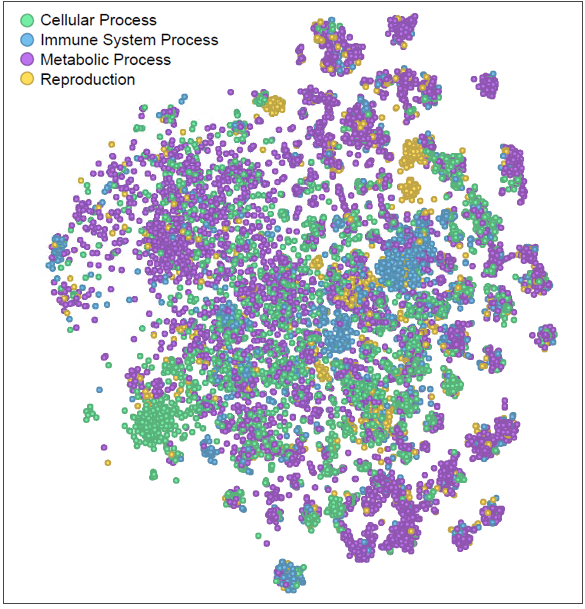
We faced a similar dilemma in gene analysis. When comparing gene lists, we counted shared genes, akin to the old NLP approach. However, genes like TLR7 and MYD88, with remarkably similar biological roles in innate immune signaling, were missed when appearing in separate lists. Just as “cat” and “kitty” were once distinct and unrecognized. In our recent Nature Communications publication [2], we draw parallels between NLP and bioinformatics, introducing Functional Representation of Gene Signatures (FRoGS) as the Word2Vec equivalent for gene analysis (Figure 2). We then harnessed the power of FRoGS to uncover compound targets. By aligning gene signatures from shRNA/cDNA perturbations with compound treatment data from the Broad’s L1000 dataset, we achieved remarkable results. Specifically, FRoGS-based AI models demonstrated a 36% recall in identifying true compound targets, surpassing the meager 9% recall achieved using the traditional one-hot encoding approach.
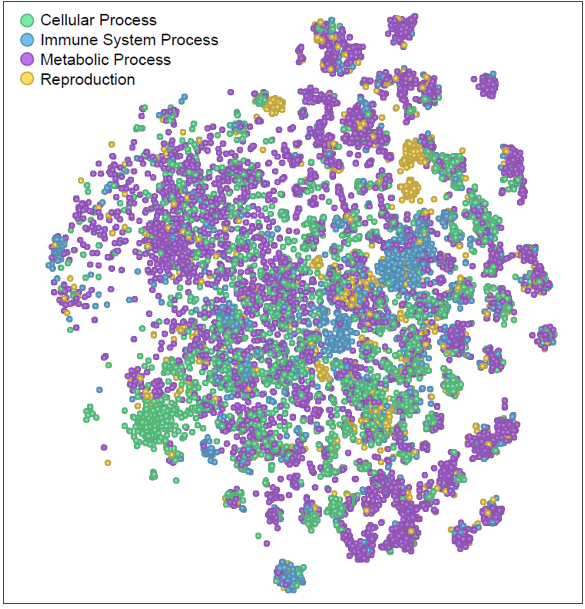
Figure 2. The t-SNE projection of the FRoGS vectors, each marker represents a unique human gene. The coloring illustrates genes with similar functions tend to form local clusters.
Why does FRoGS work? Gene functions lie at the heart of biological processes and phenotypic outcomes. When genes are naively represented as one-hot vectors, AI models must painstakingly rediscover their functions—a process demanding copious amounts of training data. Unfortunately, such extensive data may not always be available, hindering accurate predictions. FRoGS embeddings revolutionize gene representation. Each human gene is encoded as a meaningful vector, capturing both its known functions annotated in Gene Ontology (GO) [3] and its latent roles inferred from large-scale transcriptome datasets (such as ARCHS4) [4]. FRoGS enables AI models to focus on learning the intricate associations between gene functions and phenotypes without rediscovering gene functions.
In this blog we further demonstrate how FRoGS vectors enable powerful machine learning tasks, revolutionizing gene analysis beyond traditional one-hot encoding.
Example 1: Tissue-Specific Gene Expression
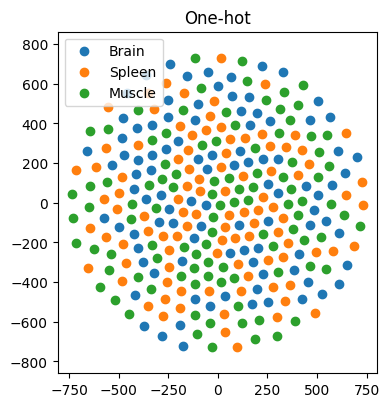
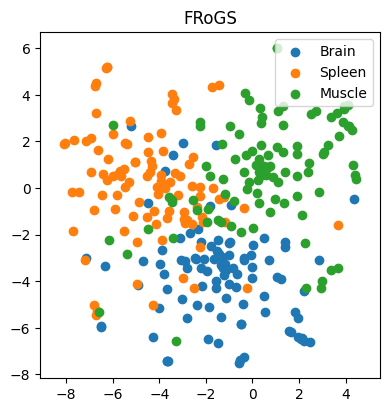
In our first example, we work with 97, 98, and 100 genes specifically expressed in the brain, spleen, and muscle, respectively. Our goal is to predict the tissue of gene expression. Traditionally, one-hot encoding places these genes in a high-dimensional space, with no inherent similarity. Consequently, AI models struggle to learn meaningful patterns, resulting in dismal classification accuracy of approximately 29% (±3%) (n = 100 simulations) — akin to random guessing (Figure 3). However, when we represent genes using FRoGS vectors, tissue-specific clusters emerge in t-SNE plots (Figure 4). The random forest accuracy soars to approximately 80% (±5%) (n = 100 simulations), demonstrating the power of FRoGS even with limited data (Figure 4).
Figure 3. The t-SNE projection of the genes using their one-hot representations. The locations of genes are random and are not suitable for machine learning.
Figure 4. The t-SNE projection of genes using their FRoGS representations. Genes with the same tissue expression patterns tend to cluster together, leading to classifiers with good accuracy.
Example 2: Gene Lists and Functional Signatures
Our dataset comprises 35, 24, and 122 gene lists associated with artery, heart, and brain, respectively. Each gene list consists of approximately 100 gene members. In the one-hot encoding approach, a gene list is represented as the sum of its constituent one-hot member gene vectors. While these gene lists exhibit some clustering patterns (Figure 5), they pale in comparison to the pronounced clusters formed by FRoGS gene signature embeddings (Figure 6). As a result, the classification accuracy for prediction is 85% (±4%) (n = 100 simulations) for one-hot vectors and 100.0% (±0.4%) (n = 100 simulations) for FRoGS vectors.
Figure 5. The t-SNE project of the gene lists using the aggregated one-hot representations of constituent gene members. Gene lists with the same tissue expression patterns tend to cluster together, leading to classifiers with good accuracy. This reflects the performance of the current gene-identity-based approach.
Figure 6. Figure 5. The t-SNE project of the gene lists using their FRoGS representations. Gene lists with the same tissue expression patterns form tight and distinct clusters, leading to classifiers with superb accuracy. This signifies the power of leveraging FRoGS for bioinformatics machine learning applications.
Conclusion
FRoGS heralds a new era in bioinformatics, akin to Word2Vec’s impact on NLP. As researchers, let’s embrace this advancement and explore FRoGS’ potential across diverse biological contexts. Visit our GitHub repository [5] and unlock the transformative capabilities of FRoGS in your next machine learning endeavor.
The Metascape forum receives many questions about how enrichment bar graphs and heatmaps are created. This blog post explains the backend algorithms.
For a given gene list, we use the accumulative hypergeometric test (or Fisher’s exact test) to compute the p-values and enrichment factors for each ontology category (referred to as a GO term or simply a term hereafter). The detailed statistics behind enrichment analysis were described previously [here]. We explained in the Metascape paper [1] that “ontology terms found in GO form a hierarchical structure of increasing granularity, making the terms inherently redundant. Terms across different ontology sources, such as GO, KEGG, and MSigDB, etc. can be closely related as well. As a result, functional enrichment analysis can identify overlapping or related terms, making it difficult to extract non-redundant and representative processes to report in the analysis output.”
Representative Terms
We cluster all enriched terms into groups and select one term from each group as the representative. For instance, the enriched terms are grouped as \(\{T_{11}^*, T_{12}, T_{13}\}\), \(\{T_{21}^*, T_{22}, T_{23}, T_{24}\}\), \(\{T_{31}^{*}, T_{32}\}, \dots\)
The first group contains three terms \(T_{11}\), \(T_{12}\), and \(T_{13}\). The group members are sorted by their p-values, where \(T_{11}\) has the smallest p-value (most negative \(\log P\)). Terms \(T_{11}\), \(T_{21}\), and \(T_{31}\) have an asterisk superscript to indicate that they are considered the best representatives of their respective groups. These asterisk terms are displayed in the bar graph and heatmap. The bar graph and heatmap contain the top 20 groups, but the plots for the top 100 groups are also available in the zip file. Results for all enriched terms are still available in the file named “Enrichment_GO/_FINAL_GO.csv” as part of the zip pakcage. This explains why term \(T_{12}\) is not found in the plots, as \(T_{11}\) is its proxy. The results for \(T_{12}\) is considered redundant to those of \(T_{11}\) , but nevertheless \(T_{12}\) can be found in “_FINAL_GO.csv”.
Excel Sheet “Enrichment”
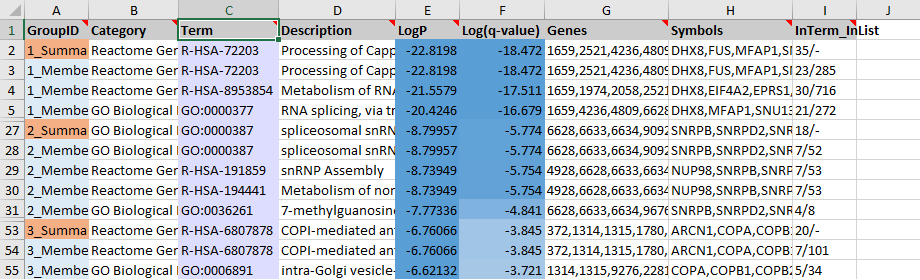
The enrichment results for the top 20 groups are also reported under the “Enrichment” sheet of the “metascape_result.xlsx” file. The top three groups are shown in the screenshot (Figure 1). The first group contains 24 members (Row 3-26, where Row 6-26 are hidden). Row 3 is the representative term for the first group, with the lowest \(Log P\) value of -22.8198 corresponding to 23 (#GeneInGOAndHitList) out of the 285 (#GeneInGO) genes. This representative entry is cloned into Row 2. Row 2 summarizes the whole group (highlighted in orange). We can see that Row 2 basically shows the same information as Row 3, except “Genes” and “Symbols” are the combination of all “Genes” and “Symbols” from Row 3 to Row 26. There are a total of 35 (#GeneInGOAndHitList) genes, therefore, we show “35” in column I. Since the summary row does not map into one specific term, there is no (#GeneInGO) count (shown as a dash in Row 2 and Column I).
Figure 1. An example Enrichment sheet. We show top three groups, where some rows are hidden to save space. The Row IDs are shown under the control column at the very left, where Row 1 contains column headers.
Clustering Algorithm
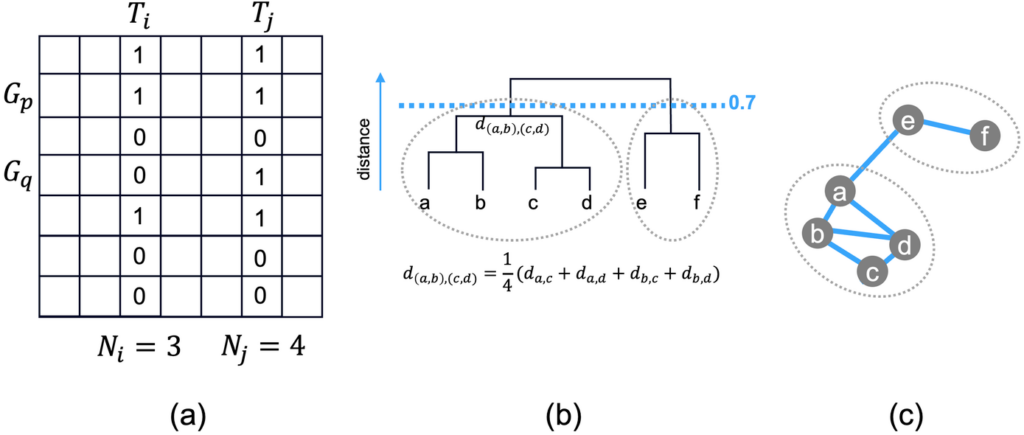
Each enriched term contains a list of genes (refer to column G in Figure 1). If we take all enriched terms as columns and all genes (from all terms) as rows, we have a binary membership matrix (Figure 2.a). We can then compute the similarity score between a pair of terms based on Kappa statistics. Some details were described by DAVID [2] and reproduced in this blog for readers’ convenience.
Figure 2.a shows an example of 7 total genes and 7 total terms. Term \(T_{i}\) has 3 genes and \(T_{j}\) has 4 genes. \(T_{i}\) and \(T_{j}\) have 6 out of 7 rows in common (3 ones and 3 zeros, \(G_{q}\) is not shared). By random chance, the probability of \(T_{i}\) and \(T_{j}\) both having the membership 1 for a given gene is \(\frac{3}{7} \times \frac{4}{7}\) and they both having membership value 0 for a given gene is \(\frac{4}{7} \times \frac{3}{7}\) . Therefore, the probability of \(T_{i}\) and \(T_{i}\) sharing the same membership value is the sum of the two, i.e., \(\frac{24}{49}\). We know that at most \(T_{i}\) and \(T_{j}\) share 100% of the rows, so we define the \(\kappa\) score as:
where the denominator is simply a normalization factor equal to the largest possible value the numerate can take. The \(\kappa\) similarity score will be a value maxed at \(1\) if two terms are identical, \(0\) if two terms are no more similar than random, and negative if two terms are even less similar compared to random. The distance between two terms is defined as \(1-\kappa\). After we compute \(\kappa\) for all pairs of terms, we can use the similarity matrix (or distance matrix) to cluster terms. DAVID invented an ad hoc fuzzy cluster algorithm [3], which we consider unnecessary. We simply run the standard hierarchical clustering algorithm with average linkage as described below.
In Figure 2.b, the hierarchical clustering algorithm first links two terms that are most similar (the smallest distance), so \(c\) and \(d\) are linked together first into tree \((c, d)\). Then \(a\) and \(b\) are linked into tree \((a, b)\), followed by tree \((e, f)\). The \((a, b)\) tree and \((c, d)\) tree are linked next, where the distance of the two subtrees is defined as:
Finally, tree \((a, b, c, d)\) is constructed, then a larger tree connect \((a, b, c, d)\) and \((e,f)\).
Figure 2. Outline of the clustering algorithm. (a) The enrichment membership matrix Gene by Term. Gene\(G_{p}\) is a member of term \(T_{i}\) and \(T_{j}\), therefore, the two cells contain 1. Gene \(G_{q}\) is not a member of \(T_{i}\), therefore, the corresponding cell contains 0.
We apply a distance cutoff of 0.7 (\(\kappa\) similarity 0.3), shown as the blue dashed line (Figure 2.b). Each subtree below the cutoff forms a group (or cluster). Therefore, \((a, b, c, d)\) form one group, and \((e, f)\) forms another group.
Due to the way distances are defined, even if \((a, b)\) and \((c, d)\) form a final group with a distance less than 0.7, it does not mean that all pairwise members have a distance below 0.7. Some distances can be higher, as long as the average is below 0.7. Assume term \(b\) has the lowest \(\log P\) among \((a, b, c, d)\); \(b\) will become the representative term for this group.
Network Visualization
Bar graphs and heatmaps only show the representative terms; otherwise, the visualization will be too busy to be useful for visual interpretation. To visualize non-representative terms, we can use a network visualization (Figure 2.c).
We show the 6 genes as nodes of a network. When a pair of nodes has a similarity \(\kappa\) higher than 0.3, they are connected by a blue edge. We see that \((a, b, c, d)\) and \((e, f)\) are grouped together respectively. However, there is no blue edge linking \(a\) and \(c\), implying \(d_{a,c} > 0.7\). Nevertheless, \(a\) and \(c\) still belong to the same group, as \((a, b)\) and \((c, d)\) have an average similarity above 0.3. Similarly, there is an edge connecting \(a\) and \(e\), even though they belong to two groups.
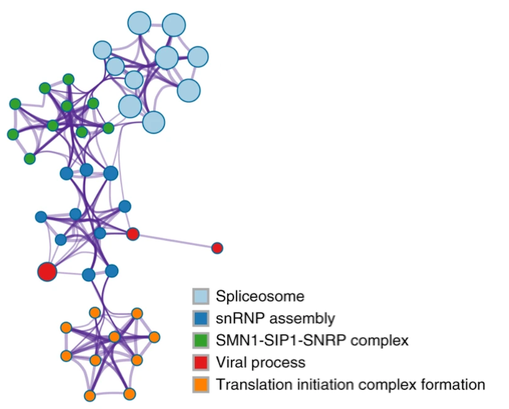
In the example network below (extracted from [4]), we see five groups consisting of densely connected members. If a group contains many members, we display at most top 10 members to keep the visualization interpretable. There are a few edges linking different clusters, indicating they are distant but related biological processes. We see that spliceosome processes are closely related to SMN1 complex, which in turn is related to snRNP assembly, which is further connected with viral processes and translation initiation complexes. To visualize individual nodes, open the “Enrichment_GO\GONetwork.cys” session file in Cytoscape.
We are excited to leverage the power of ChatGPT to improve Metascape! ChatGPT is a powerful language engine that has gained broad biological knowledge by digesting text from the Internet. GPT-4 scores a 5 in AP Biology and 99th to 100th percentile on the 2020 Semifinal USA Biology Olympiad [1]. Given appropriate prompts, ChatGPT can summarize gene functions rather impressively.
Standing on the shoulder of NCBI’s gene summary, ChatGPT describe gene functions equally well, if not better. Readers can compare the following paragraph to NCBI’s version [2] regarding gene TP53:
The TP53 gene encodes a tumor suppressor protein that responds to various cellular stresses to regulate the expression of target genes, inducing cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. The protein contains transcriptional activation, DNA binding, and oligomerization domains. Mutations in this gene are associated with various human cancers, including hereditary cancers such as Li-Fraumeni syndrome. Alternative splicing and the use of alternate promoters result in multiple transcript variants and isoforms. Additional isoforms have also been shown to result from the use of alternate translation initiation codons from identical transcript variants.
Importantly, ChatGPT summarizes disease and drug information that is critical for biomedical researchers but not yet captured by NCBI’s gene summary:
The TP53 gene is one of the most frequently mutated genes in human cancers, with mutations occurring in over 50% of all tumors. These mutations can lead to loss of function of the TP53 protein, resulting in uncontrolled cell growth and tumor formation. As a result, there has been significant interest in developing drugs that can restore TP53 function or target tumors with TP53 mutations. One example of a successful drug targeting TP53 mutations is the small molecule PRIMA-1, which has been shown to restore TP53 function in vitro and in vivo. Another promising approach is the use of gene therapy to introduce functional TP53 into tumor cells. Several clinical trials are currently underway to evaluate the safety and efficacy of these approaches in cancer patients.
ChatGPT’s ability to automate knowledge summarization becomes invaluable when little or no available human curations exist. While NCBI provides no gene summary for CCDC74A [3], ChatGPT writes:
There is limited information available on targeted drug discovery efforts for CCDC74A, as it is a relatively newly discovered gene. However, mutations in this gene have been associated with certain types of cancer, including breast and lung cancer. This suggests that targeting CCDC74A may be a potential strategy for cancer treatment. Currently, there are no drugs on the market that specifically target CCDC74A. However, there are several drugs that target other proteins involved in cell division, such as mitotic inhibitors and cyclin-dependent kinase inhibitors, which may indirectly affect CCDC74A function. Further research is needed to fully understand the role of CCDC74A in cancer and to develop targeted therapies for this gene.
and
CCDC74A is a human gene that encodes for a protein called coiled-coil domain containing 74A. This protein is involved in the regulation of cell division and is expressed in various tissues throughout the body. Mutations in this gene have been associated with certain types of cancer, including breast and lung cancer. Additionally, CCDC74A has been shown to interact with other proteins involved in cell division, suggesting that it may play a role in the development and progression of cancer. Further research is needed to fully understand the function of CCDC74A and its potential as a therapeutic target for cancer treatment.
ChatGPT’s summaries are now automatically included for all protein-coding human genes in Metascape’s Gene Annotation analyses. Two annotation columns: “Protein Functions (ChatGPT)” and “Disease & Drugs (ChatGPT)” are added to the Excel sheet after Metascape analysis. We believe this new feature will greatly assist Metascape users to review and identify gene candidates more efficiently and effectively. Just be mindful that the annotations were extracted from ChatGPT without any human curation; caution and verification will be needed, before precious time and resource is invested in further characterizing gene candidates.
We are extremely excited to make MSBio available to the bioinformatics community, including a commercial license option for for-profit entities (this post was updated on Dec 5, 2021).
Why MSBio?
Metascape was initially designed to support biologists, as we observed most gene-list analysis tools were bioinformatician-oriented rather than biologist-oriented. The reality is that the analyses implemented behind Metascape are not only difficult for biologists to perform, but also quite challenging for many bioinformaticans to implement. Frequently computational users have made inquiries regarding their desire to run Metascape analyses programmatically.
Why not provide Metascape Application Programming Interface (API), you asked? To obtain the comprehensive analysis results, Metascape utilizes computationally-expensive algorithms and visualization tools. Despite we have the best computer algorithm specialists in our team, Metascape is much more resource hungry than most other gene list enrichment services. Thus, we have to reserve our server for biologists’ use and cannot afford to expose it as the world’s shared computational hardware.
Why not release Metascape as an R package, some asked? This is not feasible, as users will not only need to install tons of software libraries (many require compilations), but also database servers, third-party tools including Cytoscape and Circos (using Perl unfortunately), etc. If we released a package, we would have been flooded with installation questions and could not breathe. There will never be a standalone MSBio installation package due to these reasons. The only alternative is to distribute MSBio almost as a preinstalled machine image. Instead of virtual machines (VM), the new technology enables such images to be delivered in the form of Docker images. We are sorry for users do not have a Docker infrastructure. Our suggestion is either convince your IT team to let you run Docker on your in-house Linux servers, or you can install Docker for your own Linux, Mac (except M1 chip), or Windows machine.
Another big hurdle for MSBio is the underlying databases. Metascape relies on over 40 databases, therefore, simply installing all Metascape code does nothing for users. As we cannot afford to have MSBio connect to our central database, we need to distribute databases with MSBio as well. First, we are not lawyers ourselves to interpret every lines of legal statements. Not all data are open for all users. Although most data providers are okay with web portals providing a nibble of their data for each analysis, as it is probably viewed as a free advertisement, redistributing the their database is certainly not in the consideration. Therefore, we need to go with a conservative minimum subset of data sources and restrict MSBio for non-commerical use (commercial users, please read on) in our licensing terms. Fortunately, most key databases such as Gene Ontology, Entrez, STRING, EggNog are free to everyone, so MSBio analyses remain rather comprehensive than most other solutions.
Non-commercial Users
MSBio is a very complex project and we are glad that we are now able to provide a convenient way for bioinformaticians to easily install the images containing both third-party tools and databases. We enabled unlimited batch analysis capability on your gene lists using your own hardware resource, while reserving our Metascape server for the users who prefers to run analysis within the browser interface. We nevertheless need to reserve the right to potentially email you, in case there is an urgent need to notifying you to stop using a certainly version due to bugs or other reasons.
The technical complexity also means that the update of MSbio will be less frequent compared to Metascape.org for the foreseeable future. We therefore request your consensus not to use Metascape as the backend for any public-facing web servers. The community needs a central free-fresh-easy Metascape.org portal. We also request your collaboration in citing the original Metascape publication in your works, as that is the only way we collect some credits for our volunteered hard work. All these terms are listed, when you register for a free MSBio license. Simply do not use MSBio, if you disagree with any.
Commercial Users
For commercial users, you should know Metascape.org keeps all analysis sessions anonymously for 72 hours max. We do not have a slight interest in your data, as we do not even have enough time to study our own data 🙂 However, we totally understand it can be a pain to convince your legal department that Metascape.org portal is safe for your proprietary gene lists. Therefore, MSBio will be a very powerful addition to your in-house bioinformatics arsenal. It empowers you to run Metascape analyses on your own hardware in parallel, without worrying about the leak of your proprietary gene lists. In addition, we can deliver the data sources we have and you have the proof for their license. We will also provide command line tools for you to export built-in Metascape ontologies, as well as appending your own in-house gene sets to enable your internal researchers to capture collaboration opportunities through Metascape analyses. Metascape.org is not for profit and all developers are volunteers, therefore, all the licensing fees will all go to support the Metascape.org servers to ensure it can continue to serve the open scientific research community for free. Please email us at metascape do team at gmail dot com to obtain an obligation-free 30-day commercial trial license.
Posted inComment, News|Comments Off on Metascape for Bioinformaticians (MSBio)
Metascape provides a rather unique protein-protein interaction (PPI) network analysis capability. In many gene list analysis resources, PPI analysis results in a rather massy hairball network. Besides stating such networks are statistically significant, there is not much biologists can say about such networks. To infer more biologically interpretable results, Metascape applies a mature complex identification algorithm called MCODE to automatically extract protein complexes embedded in such large network. Then taking advantage of Metascape’s functional enrichment analysis capability, it automatically assigns putative biological roles of each MCODE complex. Such analyses are very computational intensive and cannot be easily computed even by bioinformaticians. Regardless of its advanced PPI analysis algorithm, the results still heavily determined by the quality of its underlying PPI database.
Analyzing the publications citing Metascape, we found many users use STRING database for PPI analysis. Indeed STRING is probably the most comprehensive PPI data source, therefore, tend to provide a denser and oftentimes better looking network. The main reason Metascape has not included STRING is because we have not found a good way to cross compare STRING with other PPI data sources not yet included in STRING, especially we believe data sources such as OmniPath and InWeb_DB (the latter is no longer accessible to the public, therefore Metascape only uses an old snapshot) are presumably of higher quality than most STRING data. All interactions in STRING has a quality score, therefore, one can prioritize and use only the high-quality subset, however, we are not able to assign similar scores to interactions not yet captured by STRING. In the latest Metascape release, we now propose a way to compile an integrated PPI database including STRING, BioGrid, OmniPath and InWeb_DB. We believe this is an important step forward to significantly bring greater value of Metascape PPI analysis to our users.
Physical Interactions and Genetic Interactions
There are two types of protein-protein interactions: physical interactions and genetic interactions. “Genetic interactions capture functional relationships between genes using phenotypic readouts, while protein-protein interactions identify physical connections between gene products” [ref]. Physical interaction means two proteins are biochemically bond, either directly or through a complex. Genetic interaction more refers to functional interaction, such as regulation, so we will call them functional interactions as well. Oftentimes, genetic interactions include observations derived through computational means, therefore, they tend to be less accurate and potentially are more agreeable with Gene Ontology (therefore, less of a truly orthogonal data source). In BioGrid, these two types are counted independently [see link] and we often only use physical interactions to get results that are more conservative. Many STRING users tend to ignore the differences and apply both sets to their data, therefore, their STRING networks do appear denser. We do not believe there is a straightforward answer on either using physical only or combining both interaction types . If the physical-only network is already sufficiently dense, we should use it as it is more reliable and provides evidence more independent from the GO enrichment analysis. However, if the physical-only network is too sparse, a combined network is needed in order to gain useful biological insights.
Evidence Score for Non-STRING Data
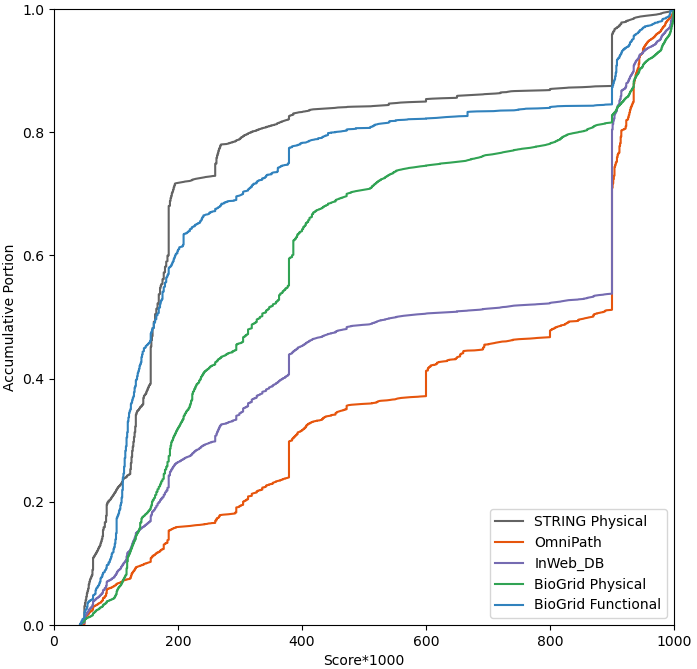
STRING provides a probabilistic framework to assign a confidence score for each PPI pair, by assuming all evidences are independent. We therefore can assign both a physical score and a combined score for its data record. But how to assign a score to data not captured in STRING, so they can be combined?
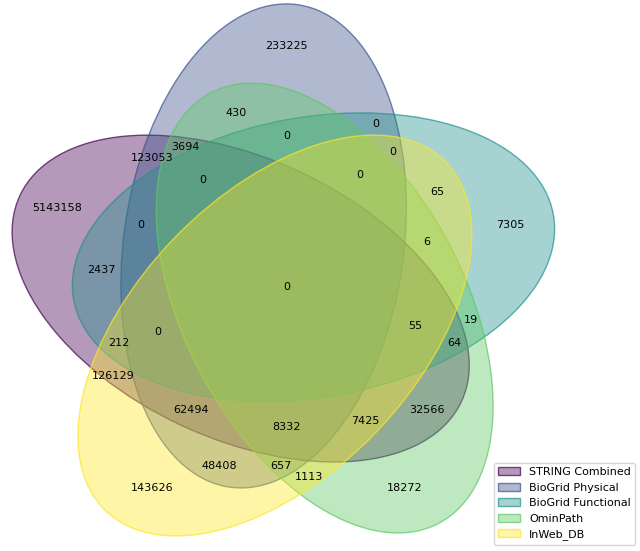
First, for those PPI pairs that are already included in STRING, we check their STRING physical scores. The figure below shows the physical score distribution of BioGrid physical subset, BioGrid functional subset, OmniPath, InWebDB and STRING physical subset itself using human data. Notice these are accumulative curves for their score distributions. We can see about 50% of the PPI data in OmniPath and InWeb_DB have a physical score > 0.9, i.e., these two data source indeed are of high quality even by their STRING physical scores. Then BioGrid physical subset has better quality than its functional subset and STRING subset has the lowest quality. I.e., in terms of data source quality, we can say OmniPath > InWeb_DB > BioGrid (Physical) > BioGrid (Functional) > STRING (Physical), in line with we expected.
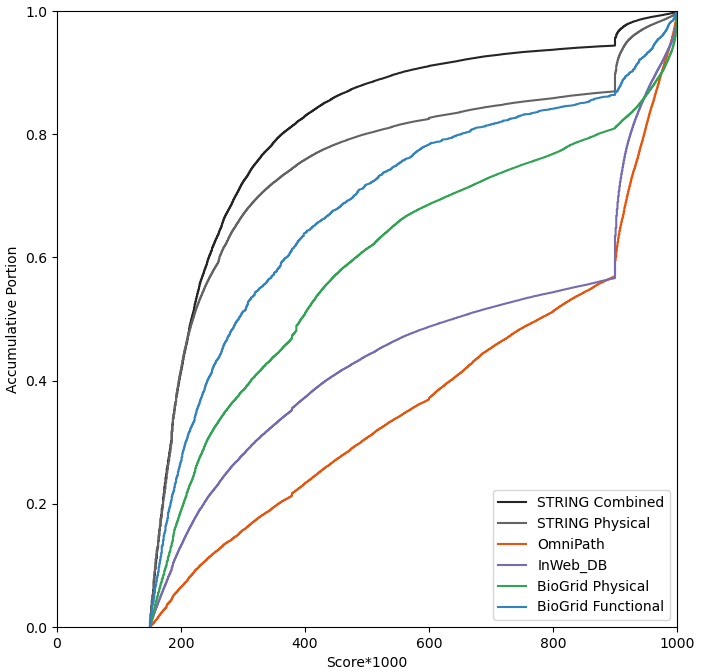
Now since we cannot assign individual STRING scores to those pairs that are not already in STRING, we can only assume all data in non-STRING data sources share the same STRING physical score. We subjectively choose the score corresponds to ~33% percentile (1/3 of the height in the accumulative curve) of the above distribution. That is we set OmniPath, InWeb_IM and BioGrid (Physical) a STRING physcial score of 0.537, 0.356, 0.260, respectively. Then we take the 33% percentile of the STRING physical distribution itself, 0.132, as the cutoff. Therefore, all physical interactions with STRING score > 0.132 are consider a reliable subset, which we call “Physical (Core)”. “Physical (Core)” include all of OmniPath, InWeb_DB, BioGrid Physical and 2/3 of STRING Physical. Then all physical interactions, regardless of their STRING scores are included in the “Physical (All)” dataset.
Similarly, if we use combined dataset, we can assigned STRING combined score of 0.537, 0.356, 0.260, 0.221 to OmniPath, InWeb_IM, BioGrid (Physical), BioGrid (Functional), respectively. We use a cutoff of 0.187, corresponding to 1/3 of the STRING Physical, to divide the combined dataset into “Combined (Core)” and “Combined (All)”, where 2/3 of STRING interactions are retained in the Core subset.
Note: Be aware that we derive these cutoffs based on human data and assume they are applicable to all organisms. Other organism contains fewer data records, therefore, we avoid making an organism-specific threshold.
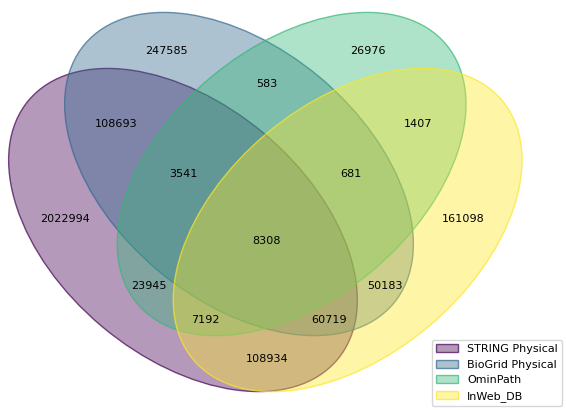
Scope of the New Database
It is exciting to report that by including STRING in Metascape, the size of our PPI database has increased significantly. Below is the Venn diagram for human, where STRING contributes >2 million new human physical PPI pairs not covered by all previous data sources.
The same goes to the size of PPI dataset, when functional data are included. The figure below shows STRING totally contributes >5 million new PPI pairs for human.
Underlying Support
For all the networks generated by Metascape, we now include an edge property called “support”. This allows users to examine the origin of each interaction pair. An example support reads like:
This is a very confidence interaction that is supported by OmniPath, InWeb_IM, BioGrid and STRING. The STRING physical score is 0.896.
Summary
We combined all data from STRING, OmniPath, InWeb_IM and BioGrid to produce four datasets: Physical (Core), Physical (All), Combined (Core), and Combined (All). OminiPath, InWeb_IM and BioGrid Physical data are consider high quality and included in all datasets. Only physical interactions are in the Physical datasets. The Core dataset contains the 2/3 of higher-scoring corresponding STRING data. Metascape “Express Analysis” defaults to “Physical (Core)” to be conservative at this point (subject to change in the future), but savvy users can choose any of the four flavors through “Custom Analysis”.
Many gene annotation, pathway, and protein interaction databases are primarily compiled for human genes/proteins. For instance, the size of the mouse interactome encompasses only ~6% of the available human interactome, even though many of these interactions are likely conserved across species. Therefore, it can be beneficial to cast gene candidates obtained in model organisms into their human orthologs prior to analysis.
In Metascape, users can choose “Analysis as Species” to designate the target organism into which the input gene list should be cast. We have been relying on NCBI’s Homologene for ortholog mapping. Homologene only covers 21 organisms, which is one of the several reasons why Metascape cannot easily support more organisms. Since Homologene does not contains P. falciparum, we have included OrthoMCL to obtain the mapping between H. sapiens and P. falciparum. It has come to our attention two years ago that Homologene appeared to became a zombie resource. If you check out the NCBI’s FTP site , the last update of Homologene was made on May 5, 2014, more than six years ago! NCBI’s response to our inquiry back in 2018 was “Homologene is in basic maintenance without update. Going forward, it is likely to be retired in the near future.” Therefore, we do need to use an alternative ortholog data source.
EggNOG is Added to Metascape
After many research and prototyping efforts, we decided to adopt EggNOG. EggNOG v5 covers more than 5000 organisms and has undergone steady upgrades every two-three years. EggNOG utilizes an phylogenetic tree to identify ortholog groups at different evolution distances. For example, if we focus on the subtree of mammals, human TLR7 protein ENSP00000370034 is uniquely linked to mouse tlr7 protein ENSMUSP00000061853. However, if we look at the tree at a very high level. TLR7 is just one of 172 human proteins related to “regulation of response to stimulus” that form an orthologous group with 167 mouse proteins. Therefore, the example orthologs of a gene depends on the evolutionary distance used, i.e., the granularity of functions one cares about. For our TLR7 example, all 172 human proteins can be many-to-many mapped to 167 mouse proteins, if we look at all organisms at very high level. To overcome this challenge, for each protein in organism A, we will identify its first-encountered orthologs in organism B be its ortholog, as we walk up the phylogenetic tree from bottom to top levels.
We then encounter another challenge. Although EggNOG is more comprehensive in scope, its mapping quality seem less desirable in many cases. For example, human KRAS ENSP00000256078 is first mapped into mouse Hras ENSMUSP00000026572. The Homologene result, linking the KRAS proteins in the two organisms, is a much more sensible result. Therefore, it seems Homologene remains a higher-quality source; we cannot simply replace Homologene with EggNOG.
Integration of Multiple Data Sources
Our current solution is to assign weights to each ortholog link: 4 for Homologene, 2 to OrthoMCL and 1 to EggNOG (the weights are very subjective). Then for the many potential orthologs for a given gene \(g_a\) in organism A, we rank ortholog candidates by their total evidence scores and pick the one with the most support. In case there is a tie, we further rank targets proteins based on the number of articles in NCBI GeneRIF and PubMed database in the descending order. The rationale is, given everything else being equal, the target protein that has been more carefully studied in the literature tend to give a better chance of providing interesting biological insights. Example: human OAS1 gene can be mapped to either Oas1a or Oas1g in mouse. Oas1a has 4 GeneRIF entries and 32 PubMed entries, where Oas1g has 0 GeneRIF entries and 17 PubMed entries. We choose Oas1a to increase the chance of better knowledgebase annotations after ortholog mapping.
Comparison of Human to Mouse Ortholog Mapping Results
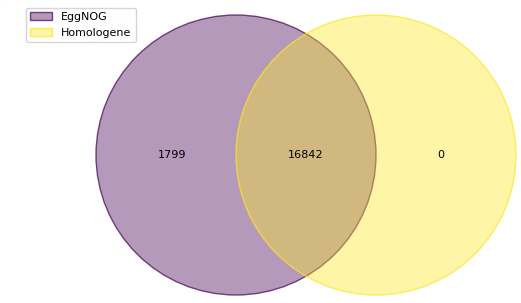
The above figure compares our new EggNOG-augmented ortholog mapping results to the previous Homologene-based results (casting from human genes to mouse genes). Our new database enables us to assign mouse orthologs to 1799 more human proteins missed by the previous Homologene-only approach.
Below are three example new pairs. Mapping CR1 to Cr2 and IFNA14 to Ifna9 make sense. Col1a1 and FLG is a suspicious link, although the proteins are functionally related. Looks like current ortholog databases still leave some room for desire.
In summary by using EggNOG in an augmented manner to improve Homologene and OrthoMCL, we have made one step forward in integrating a much better maintained ortholog data source, while we still heavily relying on a seemingly more accurate Homologene database to minimize ortholog noise.
Posted inComment, News, Ortholog|Comments Off on How Dose Metascape Compute Orthologs





Announcing Upgraded AI Gene Annotations in Metascape
In 2023, Metascape introduced AI-generated summaries for gene functions and disease implications [see previous Blog]. Powered by ChatGPT-3 at the time, these summaries served as a highly beneficial complement to NCBI’s human annotations by filling critical gaps in disease context.
In our latest database update, we are thrilled to release a completely revamped annotation dataset generated using ChatGPT-5 via Copilot. The leap in quality, scientific precision, and clinical relevance is remarkable.
To validate this upgrade, we manually reviewed multiple gene entries across the spectrum — from heavily studied genes like TP53 to lesser-known targets like CCDC74A. We also employed Google Gemini as an independent AI reviewer to compare the generations in a blind test. The result? The new version wins 100% of the time.
Please enjoy this valuable contribution of AI development to our biomedical research community. Below are two examples highlighting the dramatic improvements you can expect to see.
Example 1: TP53 (A Well-Understood Gene)
Even for highly characterized genes with mature 2023 annotations, the new version delivers significant upgrades in nuance, structure, and clinical accuracy.
The 2023 Version (ChatGPT-3)
The New Version (ChatGPT-5)
Google Gemini’s Review:
Example 2: CCDC74A (A Lesser-Understood Gene)
For newly discovered or less-characterized genes, the older model often struggled with organization and relied on vague generalizations. The new model connects structural motifs directly to functional biology.
The 2023 Version (ChatGPT-3)
The New Version (ChatGPT-5)
Google Gemini’s Review: