Requested by some users, we here provide some technical details regarding gene enrichment metrics found in Metascape analysis results.
First a few constants:
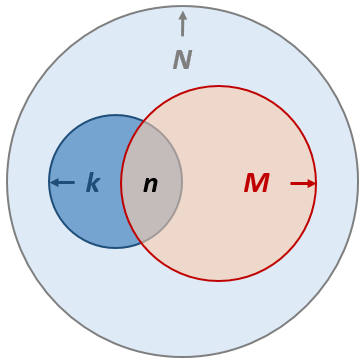
There are \(N\) total number of genes in our study pool (this is also known as the “background” gene list, defaults to all genes in the genome). A given pathway of interest consists of \(k\) gene members. Our input gene list consists of \(M\) genes, among which \(n\) are found to fall into the same given pathway.
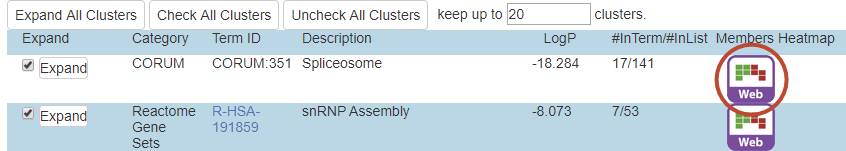
In Metascape output, we use the term “#TotalGeneInLibrary” for \(N\) (big light blue circle), “#GeneInGO” for \(k\) (small dark blue circle), “#GeneInHitList” for \(M\) (big red circle), and “#GeneInGOAndHitList” for \(n\) (the intersection between the dark blue and the red circle).
The background hit rate is \(\frac{k}{N}\) and the hit rate within our gene list is \(\frac{n}{M}\). In Metascape, hit rate \(\lambda\) is denoted as “%InGO”.
Enrichment Factor
\[\frac{\frac{n}{M}}{\frac{k}{N}} = \frac{nN}{kM},\]
indicates how many fold more given pathway members are found in our gene list compared to what would have been expected by chance. This is abbreviated as “Enrichment” in Metascape.
p-value (\( log_{10}P\))
p-value is the most used metric. If the \(M\) input genes were randomly selected from the pool of \(N\) genes, the probability of our obtaining \(n\) genes from the given pathway is :
\[{k \choose n}{ N-k \choose M-n}.\]
hint: first choose \(n\) genes from the pathway of \(k\) members, then choose the remaining \(M-n\) genes from the rest of the gene pool \(N-k\).
The above expression is also known as hypergeometric distribution. The p-value is defined as the probability of obtaining \(n\) or more pathway members, forming a cumulative hypergeometric distribution.
\[ p = \sum_{i=n}^{\min(M,K)} {i \choose n}{ N-k \choose M-i} .\]
p-values are often provided in logarithmic based ten (“LogP” in Metascape). Therefore, a more negative p-value indicates the less chance the observed enrichment is due to randomness. At Metascape, we use the following reference, simply because that is what we have been using in our own publications for years:
Zar, J.H. Biostatistical Analysis 1999 4th edn., NJ Prentice Hall, pp. 523.
q-value
If we are given one particular pathway X and asked “if the gene list is enriched in this particular X?”, the p-value would be the answer. In enrichment analysis, we are typically given Q number of pathways (or gene sets) and asked “what pathways are enriched?”. To answer that, we loop through each one of the Q pathways, repeatedly compute p-values, one per pathway (Q can be 10,000 or more). Thus, even for a randomly selected input gene list, there is still non-trivial chance to find some pathways show good p-values simply due to the large number of the pathways we query against. This is called “multiple-test” problem in statistics.
q-value, therefore, is introduced to address this issue. One way is to simply multiply p by Q:
\[q = pQ.\]
This is called Bonferroni correction. Bonferroni formula over corrects p-value, because not all Q pathways are truly statistically independent. Due to the redundant nature of the ontology knowledgebase, the effective query count Q* is a much smaller value, for which unfortunately there is no good way to estimate.
A popular alternative is called False Discovery Rate (FDR) or BH-adjusted p-value (q-value) as introduced in:
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. 1995. Journal of the Royal Statistical Society, Series B. 57 (1): 289–300.
To calculate the BH-adjustment, all p-values are sorted from small to large first. Given a p-value p at rank i, one would expect pQ pathways to be found with the same or better p-value by chance under the Bonferroni correction. Since we only observe i such pathways, the portion of our observations to be false (i.e., false discovery rate) is:
\[\min(\frac{pQ}{i}, 1).\]
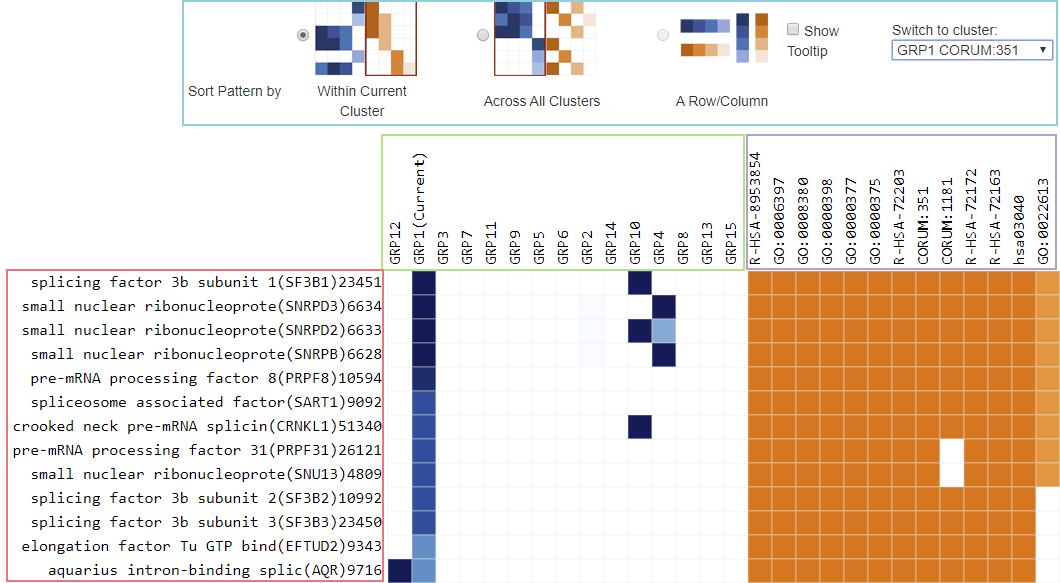
There is some additional math to ensure the resultant q-values are still in the same ascending order, but we will skip the details here. Metascape provides q-values or FDR as “Log(q-value)”.
q-value is closer to the truth conceptually, thus, sometimes reviewers may challenge you to use q-values instead of p-values. However, reviewers may not realize FDR makes use of Bonferroni correction and other assumptions that cannot be validated. For instance, the value Q can be tricky to compute, as knowledgebase is incomplete and not all ontology sources are used during the enrichment analysis. We suspect many enrichment tools may not have implemented BH correction accurately and at the end BH is just another heuristic algorithm. In practice, if the pathways of interest have p-values < \(10^{-6}\), it would remain statistically significant even under Bonferroni correction, let alone BH correction. Only when your conclusion relies on marginal p-values, such as \(10^{-3}\), you should be aware of the multiple test issue. If we view p-value or q-value as a means to rank candidate pathways for downstream validation, the ranking should remain the same.
The Excel export from Metascape provides both p-values and q-values; it meets the requirements for prestigious journals such as Cell, Nature, Science, etc.
Z-score
Z-score is correlated with p-value, we provided it within _FINAL_GO.csv file (included in the Zip package), however, non-informaticians could safely ignore this metric.
According to a Wikipedia page, on average we expect to identify \(\frac{Mk}{N}\) pathway members in our input list simply by chance. This count has a standard deviation of:
\[ \sigma = \sqrt{M\frac{k}{N}\frac{N-k}{N}\frac{N-M}{N-1}},\]
The hypergeometric distribution can often be approximated by a bionomial distribution, therefore, we can formulate a Z-score (\(Z\)-standard deviation away from the expected counts):
\[ Z = \frac{(n – \frac{Mk}{N})}{\sigma}.\]
We currently provides Z-score just for the sake of completeness, as it is just an approximate form that serves the same purpose as p-value. If users insist on using Z-score, a possible reference is (simply because this was cited in a Thomson Reuters’ Metabase document, from where we read about Z-score):
Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005 8:6:144.
Similar to the binomial approximate, Metascape estimates the standard deviation of “%InGO” (called “STDV %InGO”) as:
\[ \sqrt{\frac{\lambda (1-\lambda)}{M}} \]
Note
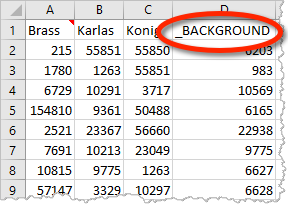
There is a tiny technical details. At Metascape, \(N\) and \(M\) actually refers to the number of genes that have ontology or gene set annotation. Those genes that have no functional annotation are excluded. This, although conceptually more rigorous, should not make a practical difference. \(N\) is default to the whole genome, users can change that by either providing a special gene list called “_BACKGROUND” in the input file, or provide it at the enrichment analysis step during Custom Analysis. If your gene pool is not based on some custom designed gene collections, you can ignore the background gene list, as the true count \(N\) is often unknown.



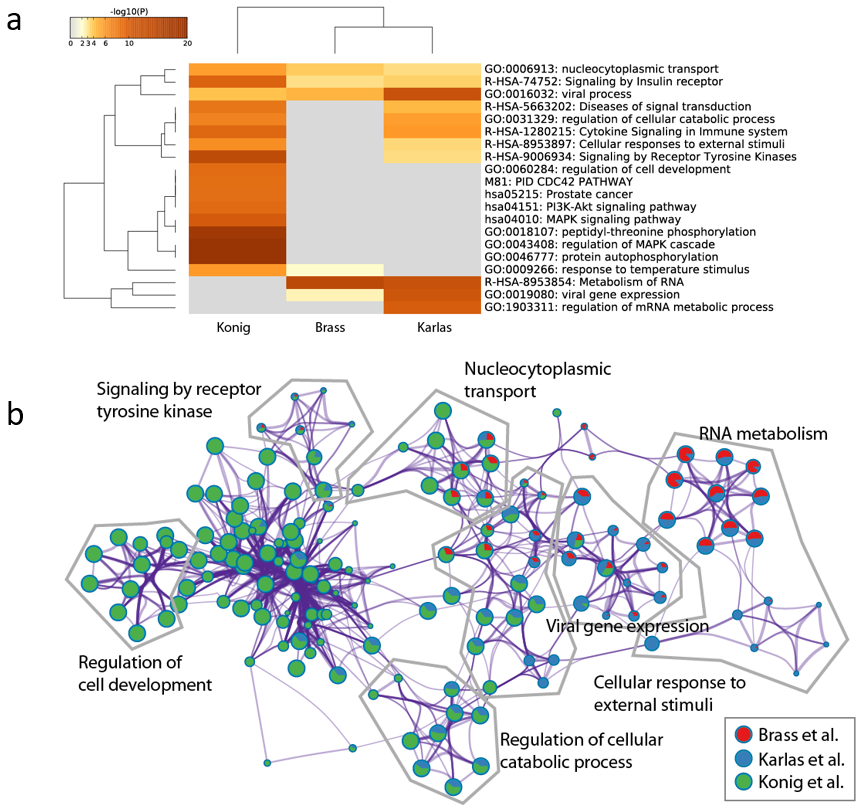
Coronascape – 为COVID-19研究特制的基因列表比较工具
简介
COVID研究者通过高通量的组学实验获取了一组基因列表后如何进行进一步的数据分析呢?高通量数据往往重复次数少,数据噪声高,想要只从自己单一的基因列表中抽取到与生物系统真实相关的分子通路就会困难一些。所以我们首先希望将信号放大,就是要找到其他已经发表或将要发表的在类似实验条件下获得的基因列表用于对照。通过比对,真实的信号得以增强,结论从统计意义上就更加可靠。目前COVID研究文献的数量在飞速增长,研究者要去阅读筛查找到相似的组学实验设计,并且对原始数据进行预处理以获得可以用于比照的基因列表其实不是一件唾手可及的事。Coronascape (http://coronascape.org) 的目的就是为了帮助COVID研究人员解决这一难题。
Coronascape是由Sanford Burnham Prebys Medical Discovery Institute, Novartis和UCSD合作共同开发的新冠病毒组学公共数据库。Coronascape收录了20篇文章360多个SARS-CoV-2相关的基因或者蛋白数据集,涵盖了七种不同的组学技术,包括转录组(RNA-Seq和scRNASeq),蛋白质组,磷酸化蛋白质组,泛素组和蛋白相互作用组。 使用Coronascape数据库可以全面深入的了解各种宿主细胞和组织中SARS-CoV-2感染后的基因表达变化,蛋白表达修饰以及相互作用关系。用户只要将自己的基因列表输入Coronascape进行Similarity Search,Coronascape会推荐数据库里相似的基因列表。当然用户也可以通过关键词搜索以获取参照组。
获取多组基因列表后,用户要通过分子通路分析和蛋白网络分析找出数据背后的分子机理,并对分析结果制成可以帮助阐明生物原理的图表。这些正是Metascape的强项,而Coronascape在后台是和Metascape无缝衔接的。Coronascape通过非常友好间的用户操作界面,让用户提交自己的基因数据与公共数据进行比较,再对生成的列表组应用成熟的系统生物学网站Metascape进行信号通路,Gene Ontology,网络分析等深入的数据分析。利用Coronascape,用户可以从单一基因列表转化为多个列表,再获取图文并茂的meta-analysis分析报告一气呵成。没有自己实验数据的用户也可以直接对Coronascape中已有的数据进行二次分析。
由于Coronascape刚刚推出,还没有发表的成果。不过已经有应用实例上传到Biorxiv (https://www.biorxiv.org/content/10.1101/2020.08.18.256776v1)。文章作者通过对比Coronascape中的数据,深入研究了SARS-COV-2的ORF9C蛋白在宿主中转录组和蛋白组等与已发表的公共数据的异同,发现ORF9C单个蛋白即可实现全病毒感染细胞和肺部组织时的免疫抑制以及细胞因子激活功能,揭示了ORF9C可能时病毒与宿主相互作用的关键蛋白之一。这是对使用Coronascape进行COV-2多重组学研究的非常好的一个示例。
下面通过一些实例介绍一下Coronascape的用法。
示例一:通过用户的基因列表从Coronascape查找相似的基因列表
1. 将您的基因列表粘贴到“User’s Gene List”中。
2. 在“Recommendation”中,单击“Similarity Search”。
3. 与用户的基因列表相比,您将能够看到按相似性排名的基因列表。您可以单击“Download Similarity”以获得详细的基因列表。
4. 您可以单击“Use Selected”或“Append Selected”以将选定的基因列表添加到“要比较的参照列表”中。
5. 添加基因列表后,单击“ Metascape Analysis”以对用户的基因列表和从Coronascape中选择的基因列表进行系统分析。
示例二:将用户的基因列表与特定公共研究的基因列表进行比较
1. 将您的基因列表粘贴到“User’s Gene List”中。
2. 在“Browse All”中,查找您感兴趣的研究。单击您要添加到参考列表的基因列表。
3. 单击“Use Selected”将基因列表添加到参考列表,或单击“Append Selected”以添加到现有基因列表。
4. 此步骤可以跳过。如果要保存选定的基因列表以备将来使用,请单击“Download Reference List”。该列表可以直接提交给Metascape进行分析。
5. 单击“ Metascape Analysis”执行系统分析。
示例三:将用户的基因列表与A549细胞系的相互作用组数据进行比较
1. 将您的基因列表粘贴到“User’s Gene List”中。
2. 在“Search”的“Technology”中,选择“ Interactome”,在“Source”中,选择“ A549”。
3. 单击“Search”以找到匹配的基因列表。
4. 单击“Use Selected”将基因列表添加到参考列表,或单击“Append Selected”添加到现有的基因列表。
5. 单击“ Metascape Analysis”执行系统分析。
示例四:对Coronascape中的基因列表执行Metascape重新分析
1. 将您的基因列表粘贴到“User’s Gene List”中
2. 使用“ Recommendation”,“ Search”,“ GO Heatmap”或“ Browse All”功能将选定的基因列表添加到“ Reference Lists to Compare”中。
3. 单击带有或不带有用户基因列表的Metascape进行系统分析。